
SESSO FETALE E COMUNI ANEUPLOIDIE SESSUALI
Esegue lo screening sulle aneuploidie cromosomiche fetali correlate agli autosomi 21, 18,13 ed ai cromosomi sessuali X,Y determinando, anche il sesso fetale che, su richiesta, può essere taciuto.
Come specificato per il test FetalDNA Cariotipo, questo è uno screening che indaga non solo sulle aneuploidie cromosomiche fetali correlate agli autosomi 21, 18, 13 ed ai cromosomi sessuali X,Y ma si spinge ad esaminare anche tutti gli altri cromosomi come avviene nello studio del classico Cariotipo Fetale.
Ogni individuo presenta 2 copie di ciascun cromosoma e con il termine aneuploidie si intendono anomalie numeriche dei cromosomi. Con il termine TRISOMIA si intende che, per quel determinato cromosoma, si osservano 3 copie, anziché 2, di quel cromosoma. Con il termine MONOSOMIA si intende che, per quel determinato cromosoma, si osserva 1 copia, anziché 2, di quel cromosoma. Le aneuploidie studiate dal FetalDNA sono le più importanti, e comuni, che possono interessare il feto.
TRISOMIA DEL CROMOSOMA 21, è l’aneuploidia più comune e si riferisce alla presenza di una copia in più del cromosoma 21. Questa sindrome è conosciuta come sindrome di Down e rappresenta, con una incidenza di circa 1/650 nati, la forma più comune di ritardo mentale;
TRISOMIA DEL CROMOSOMA 18, è la seconda aneuploidia più comune e si riferisce alla presenza di una copia in più del cromosoma 18. Questa sindrome è conosciuta come sindrome di Edwards ed è associata ad un elevato rischio di abortività. La sua incidenza si stima sia presente in circa 1/5000 nati.
TRISOMIA DEL CROMOSOMA 13, è causata da una copia in più del cromosoma 13 ed è nota anche come sindrome di Patau. E’ associata ad una elevata abortività; i neonati presentano diverse condizioni patologiche che sono spesso causa di decessi durante l’infanzia. Si stima abbia una incidenza di circa 1/16000 nati.
ANEUPLOIDIE DEI CROMOSOMI SESSUALI, sono anomalie che interessano i cromosomi sessuali XY e che possono causare nei neonati affetti difficoltà di linguaggio, motori e/o di apprendimento. La più comune di questa classe di aneuploidie è la SINDROME DI TURNER o MONOSOMIA LEGATA AL CROMOSOMA X che colpisce le donne in cui è presente una sola copia del cromosoma X ed ha una incidenza di circa 1/2700 nati. Altre aneuplodie riscontrabili con il FetalDNA sono la Trisomia del cromosoma X(XXX), la Sindrome Klinefelter e la Sindrome di Jacobs.
ANALISI DELLE ANEUPLOIDIE FETALI DI TUTTI I CROMOSOMI
Questo screening si spinge ad esaminare anche tutti gli altri cromosomi come avviene nello studio del classico Cariotipo Fetale. Esplora un gran numero di alterazioni cromosomiche determinate da riarrangiamenti strutturali ad una risoluzione di circa 10 Mb. È bene sottolineare che, a tutt’oggi, benchè esistano annunci “commerciali” diversi, in realtà nessun test NIPT sul sangue materno è in grado di ottenere risoluzioni inferiori. Per tali approfondimenti si deve ricorrere solo alla diagnosi prenatale invasiva, Amnio o Villocentesi, eseguendo uno studio specifico con microarrays.
Ogni individuo presenta 2 copie di ciascun cromosoma e con il termine aneuploidie si intendono anomalie numeriche dei cromosomi.
Grazie alla particolare elaborata valutazione bioinformatica si spinge ad esplorare TUTTE LE ALTRE ANEUPLOIDIE DI TUTTI GLI ALTRI CROMOSOMI, ispezionandone anche la struttura interna con definizione nell’ordine delle 10 megabasi.
ANALISI DELLE PIÙ FREQUENTI SINDROMI DA MICRODELEZIONE
Di seguito vengono elencate le principali sindromi da microdelezione indagate:
Sindrome di DiGeorge (delezione 22q11.2) è dovuta alla perdita (delezione) di una porzione del cromosoma 22. I bambini colpiti da sindrome di DiGeorge possono mostrare sviluppo incompleto del timo e delle ghiandole paratiroidi, cardiopatie congenite e anomalie del viso. Ha una incidenza di circa 1/3500 nati vivi.
Sindrome Cri-du-chat (delezione 5p) è dovuta dalla delezione di una regione più o meno estesa del braccio corto del cromosoma 5. E’ caratterizzata da ritardo psicomotorio, microcefalia, anomalie del volto e dall’emissione di un pianto molto tipico (simile al miagolio di un gatto) durante la prima infanzia. Si stima abbia una incidenza che varia da circa 1/15000 a 1/50000 nati vivi.
Sindrome di Prader-Willi (15q11-q13) è una patologia molto eterogenea determinata da delezioni sul cromosoma 15. E’ caratterizzata da anomalie ipotalamico-pituitarie associate a grave ipotonia nel periodo neonatale e nei primi due anni di vita disturbi comportamentali associati a problemi psichiatrici gravi e ritardo nello sviluppo psicomotorio. Ha una incidenza di circa 1/25.000 nati vivi.
Sindrome di Wolf-Hirschhorn, è dovuta a una delezione del braccio corto del cromosoma 4 (regione 4p16.3). La prevalenza è di 1:50.000 nati e interessa più spesso le femmine rispetti ai maschi (2:1) Si osservano marcato ritardo della crescita prenatale, lentezza costante nel guadagno del peso postnatale, una facies tipica ad “elmo da guerriero greco” (radice del naso larga che continua sulla fronte) molto più evidente prima della pubertà, microcefalia ed altre
anomalie facciali. Sono presenti anomalie scheletriche, come la cifosi o scoliosi con malformazione dei corpi vertebrali, costole fuse o accessorie, piedi torti e schisi delle mani.
È presente ipotonia con ipoplasia delle masse muscolari, che può associarsi a difficoltà alimentari e contribuire al ritardo della crescita. Il ritardo dello sviluppo è grave.
Sindrome di Jacobsen, che determinano anomalie congenite multiple/ritardo mentale, ed è causata dalla delezione parziale del braccio lungo del cromosoma 11. La prevalenza è stimata in circa 1/100.000 nati, con un rapporto femmine/maschi di 2:1. I segni clinici più comuni sono il ritardo di crescita pre e post-natale, il ritardo psicomotorio e caratteristici dismorfismi facciali.
Sindrome da delezione 1p36, causata da una delezione della parte distale del braccio corto del cromosoma 1. Presenta dismorfismi facciali caratteristici, ipotonia, ritardo dello sviluppo, deficit cognitivo, convulsioni, cardiopatie, sordità e ritardo della crescita a esordio prenatale È considerata una delle più comuni sindromi da delezione cromosomica, con un’incidenza di 1/5.000-10.000 nati vivi.
Sindrome di Angelman, è una malattia neurologica, di origine genetica, caratterizzata da grave ritardo mentale e dismorfismi facciali caratteristici. La sua prevalenza è stimata tra 1/10.000 e 1/20.000. I pazienti appaiono normali alla nascita. Nei primi 6 mesi di vita possono manifestarsi disturbi dell’alimentazione e ipotonia, seguiti da ritardo dello sviluppo tra i 6 mesi e i 2 anni. In genere i sintomi caratteristici della AS si manifestano a partire dal primo anno di vita, con grave ritardo mentale, assenza del linguaggio, crisi di riso associate a movimenti stereotipati delle mani, microcefalia, macrostomia, ipoplasia mascellare, prognatismo e disturbi neurologici con andatura da ‘burattino’.
Sindrome di Langer-Giedion, E’ causata dalla microdelezione del cromosoma 8q23.3-q24.13, che porta alla perdita di almeno due geni: TRPS1 e EXT1. Questa sindrome è caratterizzata da deficit cognitivo, associato a varie anomalie, compresa la cute ridondante, le esostosi cartilaginee multiple, la facies caratteristica e le epifisi falangeali ‘a cono”. La gravità e il numero di queste anomalie variano nei diversi pazienti
Sindrome di Koolen-de Vries La monosomia 17q21.31 (sindrome da microdelezione 17q21.31) è un’anomalia cromosomica caratterizzata da ritardo dello sviluppo, ipotonia nell’infanzia, dismorfismi facciali e comportamento amichevole/amabile. La prevalenza della sindrome è stimata in circa 1/16.000. I dismorfismi facciali sono caratterizzati da fronte alta e ampia, facies allungata, rime palpebrali rivolte verso l’alto, epicanto, forma anomala del naso (tubulare o a forma di pera), punta del naso globosa, orecchie grandi e prominenti e labbro inferiore anteverso. Sono frequenti le anomalie della pigmentazione e della consistenza dei capelli.
La neuropatia ereditaria con suscettibilità alle paralisi da pressione (HNPP) è una malattia ereditaria dei nervi periferici con mononeuropatia ricorrente, di solito scatenata da blande attività fisiche. In rari casi, la HNPP può esordire con una plessopatia brachiale con paralisi e perdita sensoriale dolorosa monolaterale del braccio. Di rado si osservano lievi anomalie della funzione dei nervi cranici. In alcuni casi, mancano i riflessi tendinei profondi e sono presenti piedi cavi. Il fenotipo della HNPP spesso evolve in una polineuropatia motoria sensitiva simmetrica nei pazienti anziani.
Sindrome da delezione 18q Le persone con la delezione prossimale del braccio lungo del cromosoma 18, hanno un cromosoma 18 intatto, ma l’altro è mancante di una parte di dimensioni variabili che può influenzare il loro apprendimento e sviluppo fisico. La maggior parte delle difficoltà cliniche sono probabilmente causate dalla presenza di una sola copia ( anziché due) di un certo numero di geni. Comunque, gli altri geni del bambino e la sua personalità lo aiutano a determinare il suo futuro sviluppo, i bisogni e le capacità.
Sindrome di Alagille (AGS) è caratterizzata da colestasi cronica con scarsi dotti biliari interlobulari, stenosi periferica dei rami dell’arteria polmonare, anomalie dei segmenti vertebrali, facies caratteristica, embriotoxon posteriore/anomalie del segmento anteriore, retinite pigmentosa e displasia renale. La prevalenza stimata è di circa 1/70.000. Nei neonati la malattia può manifestarsi associata a ittero prolungato con iperbilirubinemia e/o segni e sintomi cardiaci. Le anomalie cardiache includono atresia o stenosi polmonare, difetti del setto atriale e/o ventricolare, tetralogia di Fallot, dotto arterioso pervio. Le anomalie oftalmiche includono embriotoxon posteriore (75% dei casi), retinite pigmentosa, anomalie pupillari e del disco ottico. In alcuni casi si osservano ritardo della crescita e dello sviluppo e malassorbimento dei grassi (può verificarsi rachitismo). È possibile riscontrare displasia renale
Sindrome di Rubinstein-Taybi è una sindrome malformativa rara ma molto severa, caratterizzata da anomalie congenite (microcefalia, facies caratteristica, pollici e alluci larghi e ritardo della crescita postnatale), bassa statura, disabilità cognitiva e disturbi comportamentali.
Sindrome di WAGR. WAGR è l’acronimo per la sindrome tumore di Wilms – aniridia – anomalie genitourinarie – ritardo mentale. Il tumore di Wilms o nefroblastoma è il tumore renale più frequente nei bambini, responsabile del 6-8% dei cancri pediatrici. Circa l’1% di questi tumori si associa a aniridia. La prevalenza della sindrome WAGR è inferiore a 1 su 100.000 nati. La sindrome è associata a un rischio aumentato di tumore di Wilms, che si può evidenziare a qualsiasi età, e a aniridia totale o parziale, con possibile glaucoma o cataratta, disturbi genitourinari, che variano da ambiguità sessuale a ectopia dei testicoli e ritardo mentale variabile. In un bambino sono stati riportati segni oculari atipici quali microftalmia bilaterale, anomalie corneali e agenesia della camera anteriore sinistra con disfunzione retinica.
Sindrome di Potocki-Shaffer è caratterizzata da esostosi multiple, forami parietali, fontanella anteriore ampia e, occasionalmente, deficit cognitivo e lievi dismorfismi cranio-facciali. La sindrome è causata da una delezione di geni contigui sul braccio corto del cromosoma 11p11.2.
Sindrome di Miller-Dieker o lissencefalia tipo 1 da anomalie di LIS 1 o sindrome di Miller-Dieker (MDS) è una sindrome da delezione di geni contigui sul cromosoma 17p13.3, caratterizzata da lissencefalia classica (cervello liscio, con scarse cinconvoluzioni cerebrali) e segni facciali caratteristici. Possono fare parte di questa condizione altre malformazioni congenite. I bambini con MDS presentano un grave ritardo di crescita, di solito associato a epilessia e disturbi dell’alimentazione. In quasi il 100% dei pazienti sono presenti delezioni visibili o submicroscopiche della regione 17p13.3, che coinvolgono il gene LIS1.
Sindrome da delezione 1q21.1 è una malattia da delezione ricorrente, descritta di recente, caratterizzata da un quadro clinico variabile, diverso da quello osservato nella sindrome trombocitopenia-aplasia del radio (TAR). Il fenotipo clinico è estremamente variabile; i segni più comuni, ma incostanti, sono la microcefalia, il ritardo dello sviluppo o il lieve deficit cognitivo, i dismorfismi facciali, peraltro poco marcati, e le anomalie oculari. Non sono comuni le malformazioni congenite. Di rado sono stati descritti i disturbi dello spettro autistico, la schizofrenia o il deficit dell’attenzione con iperattività.
Sindrome di Kleefstra (KS) è una malattia genetica caratterizzata da deficit cognitivo, ipotonia infantile, grave ritardo del linguaggio espressivo e facies caratteristica, associati a una serie di segni clinici aggiuntivi.
Sindrome di Phelan-Mcdermid La sindrome da monosomia 22q13 (sindrome da delezione 22q13.3 o sindrome di Phelan-McDermid) è una sindrome da microdelezione cromosomica, caratterizzata da ipotonia neonatale, ritardo globale dello sviluppo, crescita accelerata o normale, linguaggio assente o gravemente ritardato e dismorfismi. La sindrome è sotto diagnosticata e la sua reale incidenza non è ancora nota. Le caratteristiche cliniche comuni comprendono le ciglia lunghe, le orecchie grandi o dismorfiche, le mani relativamente grandi, le unghie dei piedi displasiche, le sopracciglia folte, la dolicocefalia, le guance piene, il naso globoso e il mento appuntito. Il comportamento è simil-autistico con diminuzione della percezione del dolore e frequente masticazione e movimenti della bocca.
Sindrome di Smith-Magenis La SMS è una malattia sporadica da delezione 17p11.2 del gene RAI1 (retinoic acid-induced 1; 90%) o da mutazione del gene stesso (10%). Il quadro clinico
riconoscibile comprende segni craniofacciali (brachicefalia, fronte bombata, ipertelorismo, sinofria, rime palpebrali oblique verso l’alto, ipoplasia mediofacciale, faccia quadrata larga con sella nasale depressa, labbro superiore rovesciato a “tenda”, micrognatia neonatale), altre anomalie scheletriche. La prevalenza mondiale è di 1/15.000-25.000 in tutti i gruppi etnici, ma è probabile che sia sottodiagnosticata.
NB: Si ribadisce che le microdelezioni sopra riportate, benchè il test sia assolutamente preciso, verranno solo screenate senza che vi sia certezza diagnostica. Tali diagnosi, infatti, NON SONO OGGETTIVAMENTE POSSIBILI CON NESSUNA NIPT ESISTENTE. La loro esecuzione non essendo ancora approvata e riconosciuta dalle società scientifiche e dalle Linee Guida nazionali ed internazionali, deve essere considerata a solo titolo di ricerca scientifica e NON presenta valore clinico. Tuttavia, nei nostri test, tale ricerca, è risultata scientificamente affidabile. Si ribadisce ancora una volta che, per conferma o esclusione, si deve far riferimento solo ai test invasivi mediante tecnica Microarrays su materiale fetale prelevato attraverso Amnio o Villocentesi.
MALATTIE MONOGENICHE
Dopo l’isolamento del DNA fetale circolante nel sangue materno, questo viene processato mediante piattaforme di sequenziamento di ultima generazione (Next Next Generation Sequencing) allo scopo di indagare non solo riguardo l’eventuale presenza di anomalie associate al numero dei cromosomi (aneuploidie) ed alla loro struttura fine (microdelezioni/microduplicazioni), ma anche di analizzare alcune patologie associate a singoli geni (malattie monogeniche).
I disordini genetici possono essere ereditati in modo autosomico dominante (uno dei genitori è affetto), autosomico recessivo (entrambi i genitori sono portatori sani) o associato ai cromosomi sessuali. Alcuni disordini genetici hanno inoltre insorgenza de novo, cioè non trasmessi dai genitori ma prodotti in modo casuale.
Ad eccezione della beta talassemia, i disordini genetici a carattere autosomico recessivo analizzati non hanno alcun effetto sintomatico nel soggetto portatore sano; esiste per questo il rischio che due soggetti apparentemente sani possano produrre una prole affetta da un disordine di cui entrambi risultano portatori.
Nei casi di disordine genetico ad insorgenza de novo l’analisi dei genitori non può rilevare alcuna anomalia, essendo questa unicamente a carico del feto e non prevedibile.
Lo studio eseguito mediante Fetal Dna Total Screen ha lo scopo di escludere la presenza di alcuni disordini genetici selezionati in base alla gravità clinica ed all’incidenza nella popolazione.
| Gene | Disordine Genetico | Descrizione | Trasmissione |
| CFTR | Fibrosi cistica | È la più frequente malattia genetica tra i bambini Caucasici. La prevalenza in Europa è compresa tra 1/8.000 e 1/10.000 individui. La malattia è cronica e in genere progressiva, con insorgenza di solito nella prima infanzia o, più raramente, alla nascita. Gli organi più colpiti sono l’apparato respiratorio (bronchite cronica), il pancreas (insufficienza pancreatica, diabete giovanile e, a volte, pancreatite) e, più raramente, l’intestino (ostruzione stercorale) o il fegato (cirrosi), anche se possono essere interessati tutti gli organi interni. La forma più comune è caratterizzata da sintomi respiratori, problemi digestivi (steatorrea e/o costipazione) e difetti di crescita staturo-ponderale. La mortalità e la morbilità dipendono dall’entità della lesione bronco-polmonare. | La trasmissione è autosomico recessiva (entrambi i genitori devono essere portatori sani). Lo stato di portatore sano è asintomatico. |
| GJB2 | Sordità congenita | La sordità non sindromica associata a perdita totale o parziale dell’udito non è associata ad ulteriori segni e sintomi clinici. | La trasmissione è autosomico recessiva (entrambi i genitori devono essere portatori sani). Lo stato di portatore sano è asintomatico. |
| HBB | Beta talassemia | La beta talassemia (BT) è caratterizzata dal deficit (B+) o dall’assenza (B0) della sintesi delle catene della beta-globina che codificano per la proteina dell’emoglobina (Hb). La prevalenza non è nota, ma l’incidenza alla nascita della forma grave è stimata in 100.000/anno. La malattia è stata inizialmente descritta nel bacino mediterraneo. | La trasmissione è autosomico recessiva (entrambi i genitori devono essere portatori sani). |
| CYP21A2 | Iperplasia surrenalica congenita classica da deficit di 21-idrossilasi | L’iperplasia surrenalica congenita classica da deficit di 21-idrossilasi (21 OHD CAH classica) è la forma più comune di iperplasia surrenalica congenita (CAH); è caratterizzata dalla forma virilizzante semplice oppure con perdita di sale, che possono manifestarsi con ambiguità dei genitali nelle femmine e con insufficienza surrenalica in entrambi i sessi, e associarsi a disidratazione, ipoglicemia nel periodo neonatale (che può essere letale, in assenza di trattamento), e iperandrogenismo. | La trasmissione è autosomico recessiva (entrambi i genitori devono essere portatori sani). Lo stato di portatore sano è asintomatico. |
| HFE | Emocromatosi | L’emocromatosi tipo 1, nota anche come emocromatosi classica, non è una malattia rara. L’emocromatosi causa affaticamento cronico, melanodermia e gravi danni tissutali a livello del fegato, del pancreas, delle articolazioni, delle ossa, delle ghiandole endocrine, del cuore, che esitano in varie complicazioni nell’età adulta, come la fibrosi epatica (cirrosi associata al rischio di sviluppare il carcinoma epatocellulare), il diabete mellito, le artropatie, l’osteoporosi, l’ipogonadismo ipogonadotropo e l’insufficienza cardiaca. | La trasmissione è autosomico recessiva (entrambi i genitori devono essere portatori sani). Lo stato di portatore sano è asintomatico. |
| FGFR3 | Acondroplasia | L’acondroplasia è la forma più comune di condrodisplasia, caratterizzata da rizomelia, marcata lordosi lombare, brachidattilia, macrocefalia con fronte prominente e ipoplasia mediofacciale. | Generalmente ad insorgenza de novo, cioè non trasmessa dai genitori. |
| FGFR3 | Ipocondroplasia | L’ipocondroplasia è caratterizzata da bassa statura non armonica, lieve lordosi lombare ed estensione limitata delle articolazioni del gomito. | Generalmente ad insorgenza de novo, cioè non trasmessa dai genitori. |
| FGFR3 | Displasia tanatofora | La displasia tanatofora è una grave displasia scheletrica, in genere letale, a esordio prenatale, caratterizzata da micromelia, macrocefalia, torace stretto, e facies caratteristica. | Generalmente ad insorgenza de novo, cioè non trasmessa dai genitori. |
| FGFR2 | Sindrome di Apert | La sindrome di Apert è una forma di acrocefalosindattilia, un gruppo di disturbi da malformazioni congenite ereditarie, caratterizzato da craniosinostosi, ipoplasia mediofacciale, anomalie delle dita delle mani e dei piedi, e/o sindattilia. | Generalmente ad insorgenza de novo, cioè non trasmessa dai genitori. |
| FGFR2 | Sindrome di Crouzon | La sindrome di Crouzon è un disturbo genetico caratterizzato dalla fusione prematura di alcune ossa del cranio (craniosinostosi). Questa fusione precoce impedisce al cranio di crescere normalmente e influenza la forma della testa e del viso. | Generalmente ad insorgenza de novo, cioè non trasmessa dai genitori. |
| PTPN11 | Sindrome di Leopard | La sindrome LEOPARD è associata a difetti congeniti multipli, caratterizzata soprattutto da anomalie cardiache, cutanee e facciali. | Generalmente ad insorgenza de novo, cioè non trasmessa dai genitori. |
| PTPN11 SOS1 RAF1 | Sindrome di Noonan | La Sindrome di Noonan è caratterizzata da bassa statura, dismorfismi facciali tipici e difetti cardiaci congeniti. L’incidenza della SN è stimata tra 1/1.000 e 1/2.500 nati vivi. | Generalmente ad insorgenza de novo, cioè non trasmessa dai genitori. Può essere trasmessa anche con modalità utosomico dominante. |
| PAH | Fenilchetonuria | La fenilchetonuria (PKU) è la più comune malattia del metabolismo degli aminoacidi che, in mancanza di trattamento, determina ritardo mentale di grado variabile (da lieve a grave). | La trasmissione è autosomico recessiva (entrambi i genitori devono essere portatori sani). Lo stato di portatore sano è asintomatico. |
| MECP2 | Sindrome di Rett | La sindrome di Rett (RTT) è una malattia neurologica dello sviluppo, che interessa il sistema nervoso centrale. La RTT colpisce essenzialmente le femmine ed è una delle cause più comuni di deficit cognitivo grave nelle ragazze. | Le mutazioni indagate a carico del gene MECP2 si trasmettono in modo X linked dominante. |
| PKHD1 | Rene policistico autosomico recessivo | Il rene policistico autosomico recessivo (ARPKD) è una malattia ereditaria caratterizzata dallo sviluppo di cisti nei dotti collettori. Spesso si associa a un coinvolgimento epatico. | La trasmissione è autosomico recessiva (entrambi i genitori devono essere portatori sani). Lo stato di portatore sano è asintomatico. |
Si ribadisce che le anomalie monogeniche sopra riportate, benchè il test sia assolutamente preciso, verranno solo screenate senza che vi sia certezza diagnostica. Tali diagnosi, infatti, NON SONO OGGETTIVAMENTE POSSIBILI CON NESSUNA NIPT ESISTENTE. La loro esecuzione non essendo ancora approvata e riconosciuta dalle società scientifiche e dalle Linee Guida nazionali ed internazionali, deve essere considerata a solo titolo di ricerca scientifica e NON presenta valore clinico. Tuttavia, nei nostri test, tale ricerca, è risultata scientificamente affidabile. Si ribadisce ancora una volta che, per conferma o esclusione, si deve far riferimento solo ai test invasivi mediante tecnica Microarrays su materiale fetale prelevato attraverso Amnio o Villocentesi.
FIBROSI CISTICA MATERNA
La Fibrosi Cistica (FC) è una malattia ereditaria a trasmissione autosomica recessiva, cioè si eredita da entrambi i genitori portatori di un gene alterato. Per tale errore genetico si determina una alterazione delle mucosità dei vari organi. Gli organi frequentemente interessati sono il fegato, l’intestino, l’apparato riproduttivo e i polmoni dove il muco particolarmente denso porta a gravi problemi respiratori e a conseguenti infezioni.
Con questo esame viene effettuata l’analisi del gene materno attraverso uno screening chiamato di 1° livello che permette di analizzare le mutazioni più comuni e frequenti riuscendo ad identificare circa l’83% dei portatori. La frequenza stimata, nella popolazione italiana, dei portatori sani (spesso inconsapevoli di esserlo) è di 1 su 25 –30,quella dei nati affetti è di 1 su 2500 – 3000.
NB: Le mutazioni analizzate sono esclusivamente le seguenti: F508del, G542X, G551D, N1303K, R117H, W1282X, 621+1G>T, R553X, 1717-1G>A, 3849+10kbC>T, 2789+5G>A, I507del, R1162X, G85E, D1152H, 2183AA>G, 3659delC, R347P, 3120+1G>A, 3272-26A>G, R334W, 1898+1G>A, R560T, 711+1G>T, Y1092X, R1066C, R347H, 1078delT, R1158X, R117C, 1677delTA, E585X, G1244E, R352Q, 4016insT, R1066H, 2143delT, L1077P, S549R, G178R, 4382delA, 711+5G>A, D579G, Q552X, 852del22, CFTRdel22-23, CFTRdel22-24, IVS8.
Allorché fosse presente l’anomalia si dovrà procedere ad un approfondimento più completo sul feto per escludere anomalie sostenute da mutazioni genetiche rare o non diagnosticabili, sul sangue materno, nel feto.
PARTO PRETERMINE E DURATA DELLA GRAVIDANZA
Il parto pretermine è oggi la causa più frequente di mortalità e morbilità feto neonatale. Gli studi si sono concentrati per decenni sulla possibilità di individuare le gestanti a rischio di partorire prima della 37° settimana in modo da poter allestire tutta una serie di strategie terapeutiche che possano scongiurare questa severa evenienza. Per anni gli studiosi si sono concentrati sulle più diverse eziologie tra le quali quelle infettive, quelle morfologiche (malformazioni uterine), quelle da incontinenza del collo uterino sono state spesse invocate come cause principali. Purtroppo nessuna di queste è stata poi capace di giustificare il grande numero di nati prematuri che, nella maggior parte dei casi, non trova nessuna di tali ragioni per venire al mondo prima del tempo. Uno studio recentissimo su circa 50 mila donne ha svelato una causa inaspettata. Il parto pretermine insiste in una popolazione di gestanti geneticamente predisposta a tale evento.
Si tratta di uno dei più grandi studi statistici condotto esaminando oltre 15 milioni di polimorfismi genetici di suscettibilità (SNP).
Lo studio, durato molti anni, coinvolgendo oltre 50 Istituzioni Scientifiche di tutto il mondo, è stato recentemente pubblicato sul New England Journal of Medicine ed ha individuato mutazioni genetiche responsabili della durata della gravidanza e la tendenza al parto pre-termine.
L’equipe di ricerca genetica dell’Istituto ALTAMEDICA ha ricostruito il panel di tali mutazioni ed è in grado di offrire nel FetalDNA Total Screen un’analisi del rischio di predisposizione genetica al parto pre-termine. Come si può intuire si tratta di un formidabile strumento utile al ginecologo per individuare, fin dal primo trimestre di gravidanza, i soggetti a rischio e predisporre pertanto i supporti di prevenzione e terapia utili a scongiurare tale evenienza. Va comunque premesso che alla predisposizione genetica possono aggiungersi diversi altri fattori scatenanti il parto pre-termine.
Di questi si dovrà tenere conto indipendentemente dal risultato dello screening genetico.
Per maggiori informazioni vai al sito www.dnapretermtest.com
INFETTIVOLOGIA
Grazie a questa ricerca già nel primo trimestre è possibile sapere se la madre è stata infettata e presenta questi pericolosi agenti patogeni infettivi nella sua circolazione. In questo modo il medico potrà prendere le opportune decisioni sulla gestione della gravidanza avendo ben preciso il momento dell’infezione virale. Infatti troppo spesso accade che, durante la gravidanza, il titolo anticorpale di questi due pericolosi agenti infettivi, si positivizzi, lasciando nel dubbio lo specialista sull’epoca esatta di infezione materna. Questo provoca decisioni basate solo su valutazioni indirette e secondarie ad esempio cercando di capire l’epoca attraverso le date in cui le immunoglobuline “G” sono diventate maggiori delle “M” (sieroconversione) oppure sulla avidità delle immunoglobuline (valutazioni sempre approssimative).
La presenza, nel sangue materno, nel primo trimestre, dell’infezione citomegalica può determinare anomalie severe nel feto.
La infezione citomegalica materna tende ad essere asintomatica e raramente i pazienti vengono diagnosticati solo con sintomi clinici. Per la maggior parte delle infezioni, l’evidenza di sieroconversione materna (definita come una conversione da IgM negativa a positiva o un aumento di 4 volte del titolo anticorpale IgG nell’arco di 4-6 settimane) è sufficiente per confermare la diagnosi di un’infezione primaria . Tuttavia, l’accuratezza delle IgM anti-CMV materne per predire l’infezione materna primaria è complicata dal fatto che gli anticorpi IgM possono persistere per mesi o anche anni dopo l’infezione primaria, e possono anche essere trovati nel setting di riattivazione o reinfezione con un ceppo diverso di CMV.12
Un altro metodo per determinare la tempistica dell’infezione materna da CMV è misurare l’avidità dell’anticorpo, che si riferisce alla forza del legame dell’anticorpo con un antigene bersaglio. Mentre la risposta immunitaria ad un particolare antigene matura nel tempo, aumenta l’avidità. Pertanto, l’individuazione di IgG anti-CMV a bassa avidità all’inizio della gravidanza suggerisce una recente infezione acuta e può essere utilizzata per identificare le donne in gravidanza ad aumentato rischio di avere un feto infetto.4,13 Al contrario, la presenza di anticorpi ad alta avidità a 12-16 settimane di gestazione indica un’infezione passata, probabilmente prima del concepimento.
Il miglior metodo pertanto è la ricerca diretta del virus nel sangue materno (eventualmente completato con la ricerca nelle urine ). Per questo il Fetal-maternal total screen include questo esame fondamentale per la gestione della gravidanza
Per la infezione toxoplasmica materna il conoscere precocemente la presenza di una infezione nel compartimento materno permette di instaurare una adeguata terapia con un vero e assoluto razionale terapeutico.
DIAGNOSI ATROFIA MUSCOLARE SPINALE MATERNA
Il panel diagnostico del Fetal-DNA Total screen focalizza l’attenzione su questo disturbo genetico molto frequente, portato in una forma non evidente nella popolazione generale. In base alla frequenza degli individui affetti, si stima che almeno una persona su 35-50 sia portatrice del gene mutato!
Ecco perché è assolutamente obbligatorio escludere la presenza della mutazione quasi in uno dei genitori come fa il FetalDNA Total Screen.
L’atrofia muscolare spinale (SMA) è un gruppo di malattie genetiche che causano debolezza e deperimento nei muscoli volontari di neonati e bambini e, più raramente, negli adulti. È una delle condizioni genetiche più comuni che colpiscono i bambini. Si stima che un bambino su 6.000 in tutto il mondo sia nato con SMA.
In più del 95 percento dei casi, la SMA è causata dalla produzione inadeguata di una proteina chiamata proteina del motoneurone di sopravvivenza (SMN) essenziale per i motoneuroni. SMN è prodotto da SMN1 e in misura minore da SMN2. Questi geni sono sul cromosoma 5. In genere, le persone hanno due copie del gene SMN1 e fino a due copie del gene SMN2 in ciascuna delle loro cellule di motoneurone. Nelle persone con atrofia muscolare spinale, entrambe le copie del gene SMN1 sono alterate o mancanti. Avere copie aggiuntive (tre o più) del gene SMN2 sono associate a una malattia più lieve compensando parzialmente l’SMN1 mancante. Raramente, la SMA è causata da mutazioni in geni diversi da SMN (non cromosoma 5).
I motoneuroni si trovano nel corno anteriore del midollo spinale e controllano direttamente i muscoli scheletrici del corpo. Senza un’adeguata proteina SMN, i neuroni motori del midollo spinale iniziano a contrarsi e a morire. Quando ciò accade, il cervello del bambino non è in grado di controllare i muscoli volontari del corpo, specialmente quelli nelle braccia e nelle gambe e nella testa e nel collo. I muscoli iniziano a indebolirsi e a disperdersi. Ciò influisce su movimenti come camminare, gattonare, controllare la testa e il collo, deglutire e respirare.
Ci sono quattro diversi tipi di atrofia muscolare spinale. La classificazione è determinata dalle tappe dello sviluppo che il bambino ha colpito al momento dell’insorgenza della malattia. I tipi I e II sono i più comuni.
I tipi di atrofia muscolare spinale (SMA) sono:
SMA di tipo 1 (grave): questo tipo è anche chiamato malattia di Werdnig-Hoffmann. È il tipo più severo e più comune di SMA. Di solito è evidente alla nascita o nei primi mesi successivi (0-6 mesi). I sintomi includono gli arti floscio e il movimento del tronco debole. I bambini con questo tipo di solito hanno una capacità molto limitata di muoversi. Avranno anche difficoltà a nutrirsi e deglutire, a tenere la testa alta ea respirare. L’SMA di tipo 1 progredisce rapidamente, con l’indebolimento dei muscoli che porta a frequenti infezioni respiratorie e di solito la morte entro l’età di 2. I neonati con SMA di tipo 1 non possono mai sedersi.
SMA di tipo 2 (intermedio): i sintomi di solito compaiono tra i 7 ei 18 mesi. Il tasso di progressione può variare notevolmente. La malattia colpisce le gambe del bambino più delle sue braccia. I bambini con SMA tipo 2 non possono mai sopportare. Anche le infezioni respiratorie sono comuni con questo tipo di SMA. L’aspettativa di vita può variare dalla prima infanzia all’età adulta, a seconda della gravità delle condizioni del paziente.
SMA di tipo 3 (lieve): questo tipo di SMA viene anche chiamato Kugelberg-Welander o Atrofia muscolare spinale giovanile. I sintomi possono comparire per la prima volta durante un ampio intervallo di anni, da 18 mesi fino all’inizio dell’età adulta. I pazienti con SMA di tipo 3 possono stare in piedi e camminare, ma possono avere difficoltà ad alzarsi dalla posizione seduta. Possono anche manifestare debolezza muscolare lieve e sono a maggior rischio di infezioni respiratorie. La maggior parte dei pazienti con SMA di tipo 3 ha un’aspettativa di vita prossima al normale.
SMA di tipo 4 (adulto): i sintomi di questo raro tipo di SMA di solito non emergono fino alla seconda o terza decade di vita. I pazienti con SMA di tipo 4 possono camminare durante l’età adulta, ma di solito avvertono debolezza muscolare progressivamente lenta e altri sintomi tipici della SMA.
TROMBOFILIA MATERNA EREDITARIA
Benché non sia ancora possibile, ad oggi, stabilire con assoluta certezza la responsabilità della trombofilia ereditaria nella origine delle patologie della gravidanza dall’impianto (aborto) alla crescita (ritardo di sviluppo intrauterino), passando per la pre-eclampsia fino alle più gravi complicanze, si tende comunque a studiarne la natura e le attuali Linee Guida le prendono in considerazione per i più opportuni trattamenti profilattici in gravidanza e dopo il parto.
La maggior parte degli studi disponibili sono piccoli studi caso-controllo e studi di coorte assemblati in popolazioni eterogenee, sono spesso contraddittori e mostrano potenziali bias di esecuzione.
Aborto
Mentre le meta-analisi e uno studio retrospettivo di coorte hanno rivelato un’associazione tra trombofilie ereditarie e perdita di gravidanza del primo trimestre, studi prospettici di coorte non hanno trovato alcuna associazione tra trombofilie ereditarie e perdita fetale. L’Eunice Kennedy Shriver National Institute della rete di unità di medicina materno-fetale per lo sviluppo e la salute infantile ha testato donne a basso rischio con una gravidanza singleton in meno di 14 settimane di gestazione. La rete di unità di medicina materno-fetale ha identificato 134 donne che erano eterozigote per il fattore V di Leiden in 4.885 donne in gravidanza e non hanno rilevato alcun aumento dell’incidenza della perdita fetale. Risultati simili di nessun aumento del rischio di perdita del feto sono stati notati per portatori materni della mutazione del gene della protrombina G20210A.
Pre-eclampsia
Alcuni studi clinici hanno riportato un legame tra il fattore V Leiden e la pre-eclampsia, la preeclampsia grave e la preeclampsia prima delle 37 settimane di gestazione. Tuttavia, molti altri studi caso-controllo non sono riusciti a dimostrare un’associazione tra la mutazione del fattore V di Leiden e la preeclampsia. Diversi studi non sono riusciti a stabilire un legame tra la mutazione della protrombina G20210A e la preeclampsia o la preeclampsia grave. Diverse meta-analisi hanno suggerito un’associazione tra proteina C e deficit di proteina S e preeclampsia; tuttavia, queste conclusioni si basano su un numero limitato di studi che contenevano anche un piccolo numero di partecipanti (46). Non ci sono prove sufficienti per concludere che le trombofilie ereditarie siano associate a un aumento della preeclampsia.
Ritardo di crescita intrauterino
Diversi studi caso-controllo, coorte e revisione sistematica non sono riusciti a rilevare un’associazione significativa tra il fattore V Leiden e la restrizione di crescita intrauterina (IUGR) inferiore al 10 ° percentile o inferiore al 5 ° percentile. Una simile mancanza di associazione è stata osservata tra la mutazione della protrombina G20210A e la IUGR. Uno studio caso-controllo su 493 neonati con IUGR e 472 controlli abbinati non ha trovato alcuna associazione tra IUGR e fattore V Leiden, mutazione della protrombina G20210A, o dell’MTHFR (C677C).
Distacco di placenta.
Nel complesso, non vi sono prove sufficienti per stabilire un legame tra trombofilie e distacco della placenta. Le analisi prospettiche di coorte del fattore V Leiden, la protrombina G20210A e l’esito della gravidanza non hanno trovato alcuna associazione con la distacco della placenta. Tuttavia, una meta-analisi di studi caso-controllo ha riportato un’associazione tra distacco della placenta e sia omozigosi ed eterozigosi per la mutazione del fattore V di Leiden e un legame tra eterozigosi della protrombina G20210A e distacco della placenta. Lo studio Hordaland Homocysteine ha trovato un’associazione tra distacco della placenta e iperomocisteinemia superiore a 15 micromol / L, ma un’associazione minima tra omozigosità per il polimorfismo C677T MTHFR e distacco della placenta.
TRATTAMENTO
Appropriata gestione intrapartum per i pazienti trombofilici.
L’uso di stivali a compressione pneumatica o calze elastiche deve essere preso in considerazione per i pazienti con una trombofilia conosciuta fino a quando non sono ambulatorialmente post-partum. Inoltre, la profilassi intrapartum con eparina non frazionata deve essere presa in considerazione nei pazienti a più alto rischio. Indipendentemente dal fatto che il paziente stia ricevendo dosi profilattiche, intermedie o terapeutiche di LMWH, si dovrebbe prendere in considerazione la possibilità di sostituire una dose paragonabile di eparina non frazionata a 36 settimane di gestazione per consentire l’induzione dell’anestesia neuroassiale durante il travaglio e il parto. In alternativa, l’LMWH sottocutanea a dosi regolate o l’eparina non frazionata può essere interrotta 24-36 ore prima dell’induzione del travaglio o del parto cesareo programmato per evitare l’effetto anticoagulante durante il parto. I pazienti sottoposti a terapia anticoagulante profilattica devono essere istruiti a trattenere le iniezioni all’inizio del travaglio. Se il parto vaginale o cesareo si verifica più di 4 ore dopo una dose profilattica di eparina non frazionata, il paziente non è a rischio significativo di complicanze emorragiche. Oltre 12 ore dopo una dose profilattica o 24 ore dopo una dose terapeutica di LMWH, l’anestesia spinale non deve essere sospesa perché il rischio di sanguinamento correlato alla procedura è limitato (59, 60). I pazienti che ricevono eparina non frazionata o LMWH che richiedono una rapida inversione dell’effetto anticoagulante per il parto possono essere trattati con protamina solfato (61). Inoltre, i concentrati di antitrombina possono essere usati in pazienti con deficienza di antitrombina nel periodo peripartum.
Gestione appropriata dei pazienti trombofilici che necessitano di anticoagulazione post-partum.
Le dosi postparto di eparina non frazionata o LMWH devono essere uguali o maggiori rispetto alla terapia antepartum. L’eparina non frazionata o LMWH può essere riavviata 4-6 ore dopo il parto vaginale o 6-12 ore dopo il parto cesareo. I pazienti che verranno trattati con warfarin possono iniziare la terapia immediatamente dopo il parto. La dose iniziale di warfarin deve essere di 5 mg al giorno per 2 giorni, con dosi successive determinate monitorando il rapporto normalizzato internazionale. Per evitare la trombosi paradossa e la necrosi cutanea dal precedente effetto antiproteico C del warfarin, le donne devono essere sottoposte a dosi terapeutiche di eparina non frazionata o LMWH per 5 giorni e fino a quando il rapporto internazionale normalizzato è terapeutico (2,0-3,0) per 2 giorni consecutivi. Poiché il warfarin, l’LMWH e l’eparina non frazionata non si accumulano nel latte materno e non inducono un effetto anticoagulante nel bambino, questi anticoagulanti sono compatibili con l’allattamento al seno
Per una più completa conoscenza del trattamento delle trombofilie in gravidanza è bene rifarsi alle Linee-Guida del Royal College Obstetricians and Gynecologist :
Reducing the Risk of Venous Thromboembolism during Pregnancy and the Puerperium Green-top Guideline No. 37a April 2015 (https://www.rcog.org.uk/globalassets/documents/guidelines/gtg-37a.pdf)
MARKER BIOCHIMICI PER LA PREDIZIONE DI PRE-ECLAMPSIA
La preeclampsia si verifica nel 2-5% delle gravidanze in Occidente, ma complica fino al 10% delle gravidanze nei paesi in via di sviluppo, dove le cure d’emergenza sono spesso inadeguate o carenti. Pertanto, abbiamo bisogno di un test ampiamente applicabile e accessibile che possa consentire una diagnosi presintomatica al fine di identificare e monitorare i pazienti a rischio e fornire così la migliore assistenza prenatale per queste donne e il loro bambino. Tale test sarebbe anche utile per confermare una diagnosi clinica confondente e per studi futuri che studiano trattamenti profilattici o terapie temporizzanti.
Per essere efficace, un test di screening deve essere sufficientemente sensibile e specifico e deve fornire un adeguato valore predittivo positivo. Oggi sono stati descritti diversi indicatori promettenti, da soli o in combinazione, che potrebbero soddisfare questi criteri. Tuttavia, questi dati provenivano spesso da piccoli casi di studio con popolazioni selezionate.
Siccome oggi si ritiene che la pre-eclampsia sia la conseguenza di uno squilibrio nelle proteine angiogeniche e anti-angiogeniche placentari, l’attenzione si è concentrata nel ricercare, fin dall’inizio della gravidanza, l’esistenza di alterazioni delle proteine che, più di altre, regolano i fenomeni angiogenetici placentari. Recenti studi si sono concentrati sullo studio delle gravidanze che presentano alle cliniche specializzate con segni di disturbi ipertensivi con l’obiettivo di identificare il sottogruppo che si svilupperà una grave preeclampsia richiedono un parto anticipato entro le 1-4 settimane successive. In tali gravidanze ad alto rischio, la misurazione del siero PlGF e PAPP-A è estremamente precisa nell’identificare il gruppo target.
Il fattore di crescita placentare (PIGF) è una glicoproteina dimerica glicosilata, che fa parte della sottofamiglia del fattore di crescita endoteliale vascolare. PlGF è sintetizzato in citotrofoblasto villare ed extravillare ed ha sia funzioni vasculogenetiche che angiogenetiche. Si ritiene che contribuisca a un cambiamento nell’angiogenesi da una ramificazione a un fenotipo non ramificato che controlla l’espansione della rete capillare. Le sue abilità angiogenetiche sono state ipotizzate per giocare un ruolo nella normale gravidanza, e cambiamenti nei livelli di PlGF o del suo recettore inibitorio sono stati implicati nello sviluppo di PE.
La preeclampsia è associata a una ridotta produzione placentare di PlGF ( FIG1) e diversi studi hanno riportato che durante la fase clinica della pre-eclampsia, la concentrazione plasmatica di siero materno è ridotta. Questi ridotti livelli di PIGF sierico precedono l’esordio clinico della malattia e sono evidenti da entrambi i secondi trimestri di gravidanza. Nei test biochimici, è necessario effettuare attente regolazioni nella concentrazione misurata metabolita siero-materno per correggere alcune caratteristiche materne e gravidanza, la concentrazione sierica metabolita viene quindi espressa in un multiplo della mediana prevista (MoM) del normale.
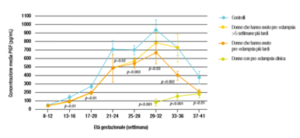
Fig. 1
Primo trimestre di concentrazioni sieriche materne di PAPP-A (Pregnancy-associated plasma protein A) e PlGF hanno dimostrato di essere modificate da età gestazionale, peso materno, razza, fumo di sigaretta, nulliparità e diabete mellito preesistente. La PAPP-A ha mostrato soprattutto valori aumentati nella sindrome di Down (FIG 2). Inoltre, il siero PlGF è anche influenzato dall’età materna. Di conseguenza, le concentrazioni misurate di PAPP-A e PlGF devono essere adeguate a queste variabili prima di confrontare i risultati con gravidanze patologiche. I valori MoM di PAPP-A e PlGF sono significativamente ridotti a 11-13 settimane di gestazione nelle donne che successivamente sviluppano la pre-eclampsia. Esiste una significativa correlazione lineare positiva tra i valori MoM di questi marcatori biochimici con età gestazionale alla consegna. Questa osservazione ulteriore conferma che la pre-eclampsia è un’entità fisiopatologica con un ampio spettro di gravità.
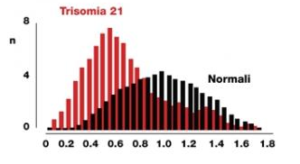
FIG 2
I valori biochimici riscontrati possono essere valutati da soli come indice di rischio di pre-eclampsia o associati ad altri parametri clinici e biofisici (FIG 3)
| FIG 3: Calcolo combinato rischio preeclampsia | |
| Epoca di valutazione | Da 11 a 14 settimane (biochimica) Da 45 ad 84 mm di cRL per ecografia |
| Biomarkers | PAPP-A e PLGF |
| Esame biofisico | Pressione arteriosa media Indice di pulsatilità delle arterie uterine |
Quando il ginecologo curante esegue una valutazione combinata dei vari parametri, sottoponendo i dati rilevati ad una analisi computerizzata mediante software dedicato (Prenatal Screening Software B·R·A·H·M·S Fast Screen pre I plus) i risultati divengono più affidabili (FIG4).

